


强关联分子体系(如固氮酶铁硫[nFe-mS]团簇)中存在大量d/f电子,其价态、自旋以及分子构型的变化具有很强的多样性,在催化反应(如生物固氮)中起着无可替代的作用。其中,固氮酶铁硫团簇目前是被认为是经典计算化学能够在高精度波函数级别计算模拟的极限,是量子计算未来可能发挥优势的实际重要体系之一。
方维海院士团队李振东老师课题组长期致力于发展针对强关联体系的经典与量子计算化学理论方法。近年来,课题组结合新一代人工智能硬件(GPU),发展了适合分布式GPU的高性能并行密度矩阵重正化群(DMRG)算法[J. Chem. Theory Comput. 20, 775-786 (2024)],开发了新的从头算DMRG程序FOCUS。利用这一新计算工具,对固氮酶中P团簇(73个活性轨道,114个电子)实现了高精度计算,能量误差逼近化学精度(1kcal/mol)。此外,课题组结合神经网络,发展了新的张量网络和神经网络混合量子态架构[J. Chem. Theory Comput. 21, 10252-10262 (2025)],开发了高效从头算神经网络量子态程序PyNQS。以上工作推进了经典计算模拟的精度和规模,有助于界定量子与经典计算化学的边界。
除了基于张量网络与神经网络的经典计算方法,课题组还致力于探索利用量子计算求解化学中的量子多体问题[J. Phys. Chem. Lett. 15, 28, 7244–7253 (2024); Precis. Chem. 3, 9, 541–553 (2025)]。近期,课题组提出了纠缠最小化轨道(EMO)的概念,以及利用自旋匹配的低键维矩阵乘积态(MPS)高效优化EMO的算法。使用这一轨道可产生更加紧凑的波函数表示,从而显著降低复杂体系初态制备所需的量子资源(见图1)。这一研究成果将为构建更高效的量子计算化学算法提供帮助。
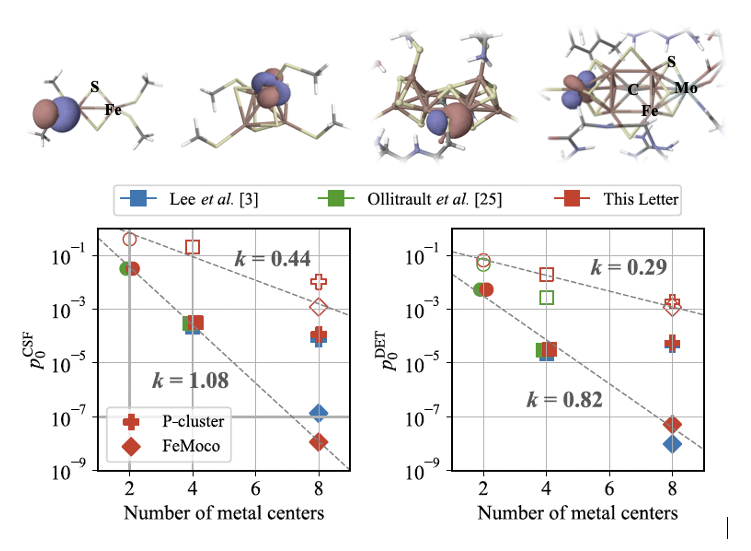
图1 纠缠最小化轨道(EMO)显著增强铁硫团簇波函数的紧凑性。
这项研究近期以“Entanglement-Minimized Orbitals Enable Faster Quantum Simulation of Molecules”为题发表于《Physics Review Letter》[Phys. Rev. Lett. 135, 210601 (2025)]。该工作得到了 “量子通信与量子计算机”国家科技重大专项经费资助。