N2O在半导体表面的光解离和光热协同解离的理论探索
——光(热)解离难辨明,共振时间是法门
光催化和光伏太阳能电池是实现“双碳”目标的重要手段,两者都涉及到光生电子-空穴对的界面分离、弛豫和复合等超快的非平衡非绝热过程。金属氧化物由于其突出均衡的物理化学性质,成为光催化和太阳能电池(燃料敏化、钙钛矿)传统的重要材料。世界上众多实验课题组采用先进的超快光谱技术,测量这些复杂体系的光生载流子动力学,获得了大量的实验数据。然而,对这些实验数据和现象背后深层次物理机制的理解却相对滞后。组合含时密度泛函理论和面跳跃方法混合量子-经典动力学较好地实现了计算效率和准确性之间的平衡,是目前处理大尺度凝聚相体系光生载流子动力学的有效工具。我们在国际上较早地开展了金属氧化物和电子给体之间的界面电荷分离的理论研究,阐释了超快电子转移受给体-受体间相互作用支配的物理机制(J. Am. Chem. Soc. 2011, 133, 19240-19249; ibid, 2012, 134, 14238-14248; Nano Lett. 2017, 17, 4038-4046);提出了表面等离激元协助的电子转移机制(J. Am. Chem. Soc. 2014, 136, 4343-4354),并被发表在Science的实验证实,随后建立了表面等离子激元协助的传统电子转移机制实现高光电转换效率的微观机理(Chem 2018, 4, 11112-11127);建立调控界面电子转移机制的新策略(J. Am. Chem. Soc. 2017, 139, 2619-2629),降低非辐射电荷和能量损失。
较强的电声耦合使得金属氧化物中普遍存在小极化子,它对光催化和光伏的影响通常是负面的,或降低寿命(J. Phys. Chem. Lett. 2021, 12, 3514-3521),或降低载流子迁移率(J. Phys. Chem. Lett. 2022, 13, 5571-5580),或兼而有之,限制了器件的转换效率。然而,鲜有理论工作系统的研究金属氧化物中小极化子的形成、跳跃、湮灭及参加化学反应。我们提出了一种简便的小极化子形成时间的计算方案,并首次在α-Fe2O3中同时构造了局域的电子极化子和空穴极化子,获得了与实验测量一致的逾百皮秒寿命的电子极化子(npj Comput. Mater. 2022, 8, 148),阐释了掺杂提升极化子跳跃的机制,并提出通过自旋调控(JACS Au 2021, 1, 550-559)和分子调控(JACS Au 2022, 2, 234-245)规避极化子的不利影响,促进光化学反应(ACS Catal. 2022, 12, 6702-6711)。众所周知,光化学反应中必然包含热效应,如何分辨光化学反应是纯粹的光催化机制还是光热协同催化机制一直是实验研究和理论研究关注的热点和难点。深刻理解这一科学问题,有助于高效光催化材料和光伏材料的合成与制备。
为了回答这一问题,北京师范大学龙闰教授和方维海院士课题选取N2O和还原的TiO2(110)表面为模型体系(此时极化子态提供了TiO2的可见光光催化活性),采用非绝热分子动力学模拟结合“Impulsive Two-State”方法,从时域和能量范畴在原子尺度对N2O光解离反应过程机理进行了详细的研究。
当N2O接收TiO2导带光激发电子,瞬时形成N2O-阴离子自由基,我们定义N2O-共振寿命为考察参数,探讨了N2O光催化解离过程中的光解效应和光热协同效应。如图1所示,当较短或者中等时,光热协同分解机制主导解离;当较长时,光分解支配解离过程。
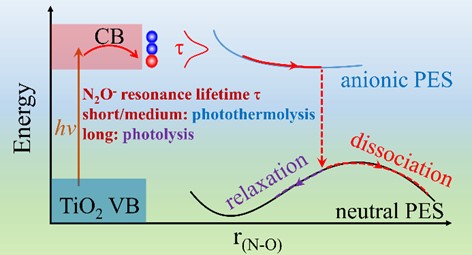
图1. TiO2光催化分解N2O示意图,N2O-共振寿命决定了反应的机理。
如图2所示,共振寿命决定了解离的N2O难易程度和解离机制。这里仅以氧端连接在TiO2(110)表面的模型进行讨论,氮端连接体系的讨论请见原文。随着N2O-共振寿命的增加(1-4 fs),N2O弯曲振动模式和N-O对称伸缩振动模式被逐渐被激活,使得NNO从~180逐渐降到~150,N-O逐渐拉长,直至断裂。N2O解离的时间从 = 2 fs时的~1800 fs缩短至 = 4 fs时的~70 fs,而 = 1 fs,光子和热能给予的动量还不能克服N-O解离能垒,因此分子的键长和键角在各自平衡位置发生周期性振荡。=2fs时从头算分子动力学(AIMD)的几个代表性结构也证实了N2O弯曲振动模式和N-O对称伸缩振动模式被逐渐活化。
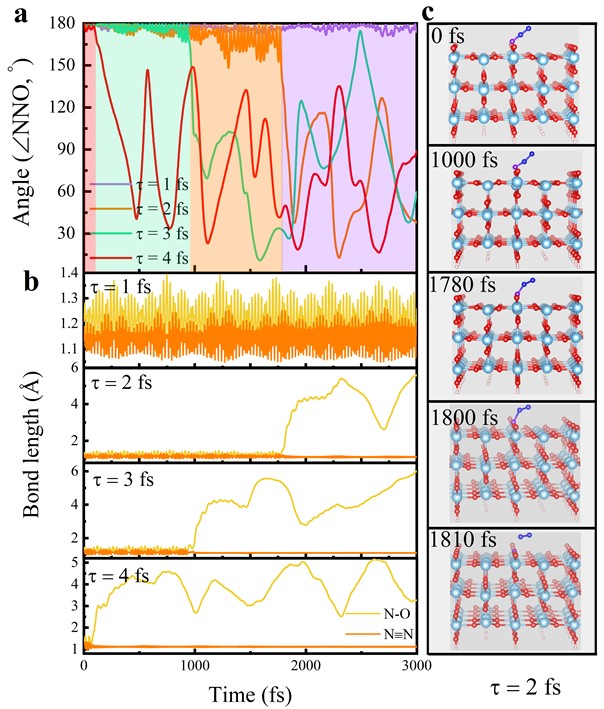
图2. 不同N2O-共振寿命下NNO 和N-O/ N≡N键长演化(左图)和 = 2 fs的几个代表性几何结构演化。
电子结构计算(图3)给出两个重要结论。一,态密度计算表明随着阴离子的共振寿命增加,N2O的LUMO逐渐向TiO2的导带底移动,有利于接受TiO2导带的光生电子,活化两种振动模式;二,晶体轨道布局分析表明,当逐渐增加,费米能级之下有越来越多的电子占据,N-O键的强度也逐渐减弱。电子结构分析表明较长的共振寿命有利于更多的电子转移给N2O分子,从而促进N-O键的活化,更有利于N2O解离。
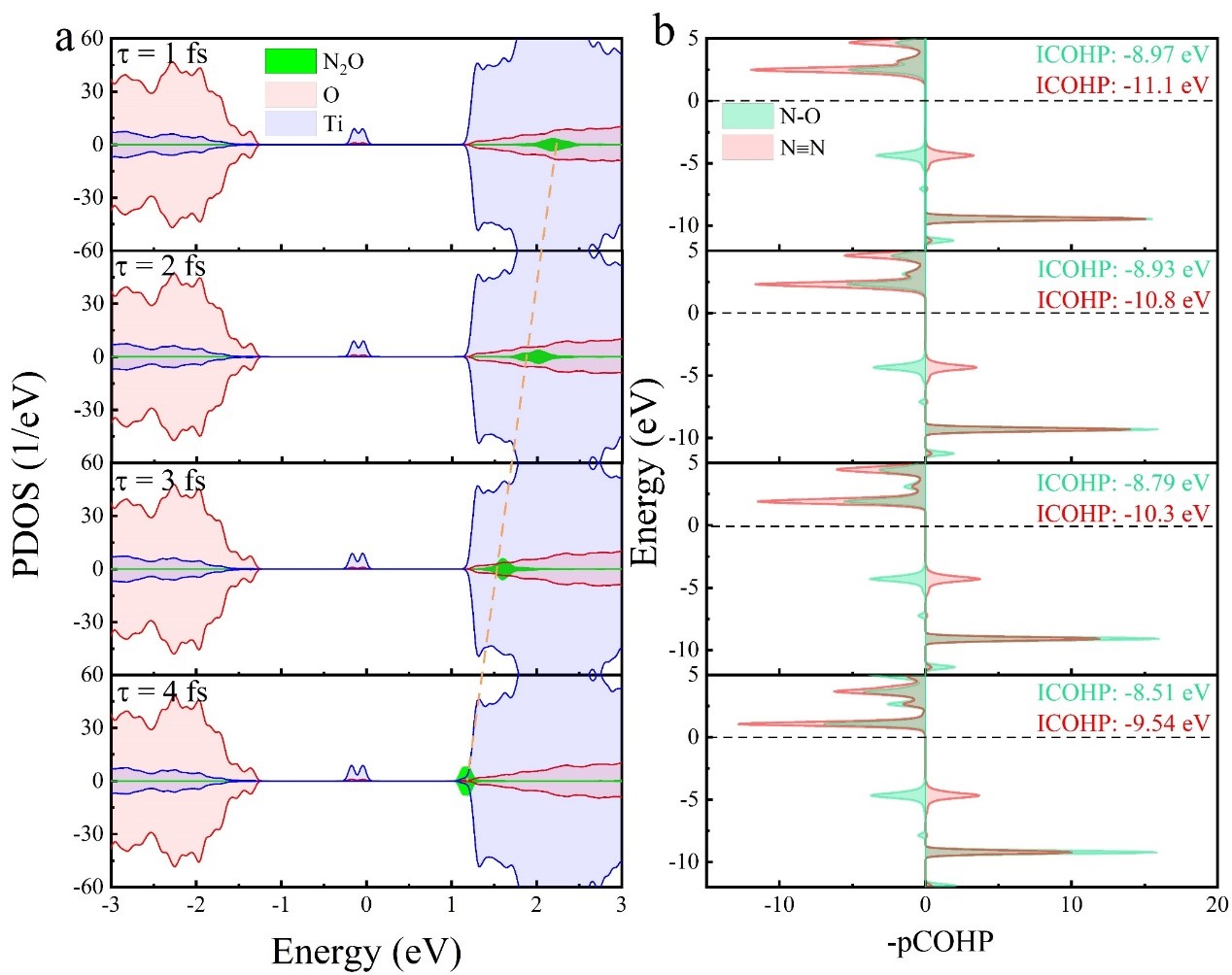
图3. 不同N2O-共振寿命的态密度及化学键强度分析。
静态电子结构计算和AIMD模拟给出的初步结果激励了随后的非绝热动力学模拟,用以分析N2O-阴离子的形成时间(N2O俘获TiO2导带电子时间)和存活时间(电子-空穴复合时间),从而确定N2O分子是否能够真正分解及分解的机理为何。依据实验,我们考虑了TiO2的带边激发和从靠近导带底的小极化子态(trap2)的激发,图4a显示N2O LUMO俘获TiO2导带电子的时间尺度分别为45fs和74 fs(N2O-阴离子的形成时间),而对应的能量弛豫时间为129和133 fs,大约是电子转移的2倍,表明光激发电子可以有效地被N2O分子有效提取。时间演化的N2O LUMO如图4b所示,当 = 1 fs,N2O LUMO一直位于TiO2导带内,并发生周期性的振荡。当 = 2- fs时,N2O LUMO在初始时刻发生振荡,但在获得足够的局部晶格热能后N2O LUMO逐渐转移至TiO2导带底,随后接受TiO2导带光激发的热电子形成新的N2O-阴离子,紧接着经过约50 fs的热活化使得分子发生解离。非绝热分子动力学模拟计算了新形成的N2O-阴离子上电子与空穴(trap2)的复合时间大约为700 fs,远大于新形成的N2O-稳定在TiO2导带底并发生解离需要的时间(~70 fs),表明光热协助解离可以有效地发生。
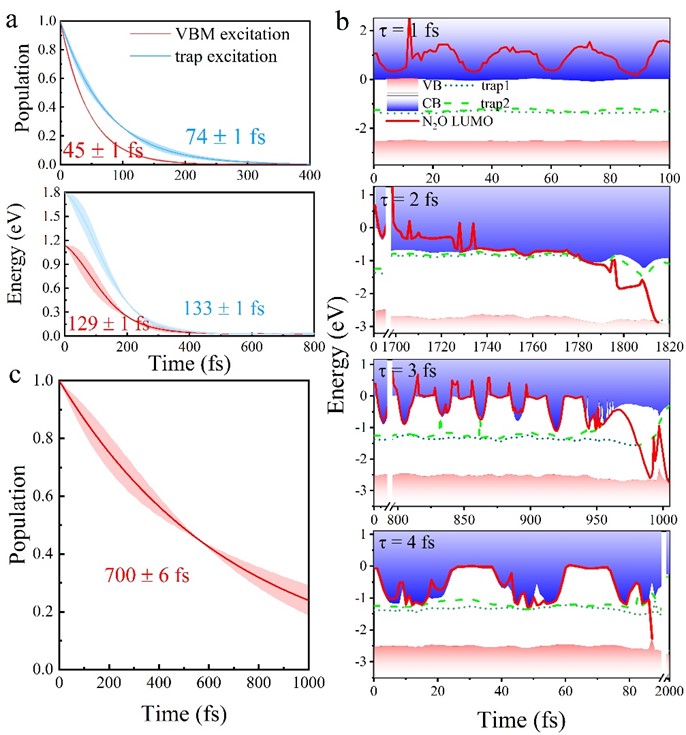
图4. 形成N2O-涉及的电子转移和能量弛豫、N2O-电子-空穴复合及N2O LUMO随时间的演化。
根据电子结构计算、AIMD和非绝热动力学模拟结果,我们提出了详细的解离机理,如图5所示。当N2O-寿命较短时( = 1 fs), 光子和热能无法完全激活N2O的弯曲振动模式和N-O的伸缩振动模式,分子不能解离。当 = 2-4 fs时,光子给予N2O的动量部分激活了上述两种振动模式,在几十至上千飞秒的热波动的协同作用下,充分激活两种振动模式,使得N2O LUMO转移至TiO2的导带底,N2O可以重新捕获光激发热电子形成新的N2O-阴离子,然后经过小于100 fs的热波动使得分子克服解离能垒发生解离,进行光热协助的解离过程。当 ~20 fs时,分子可以被光子给予的动量直接解离,光解离是唯一过程。
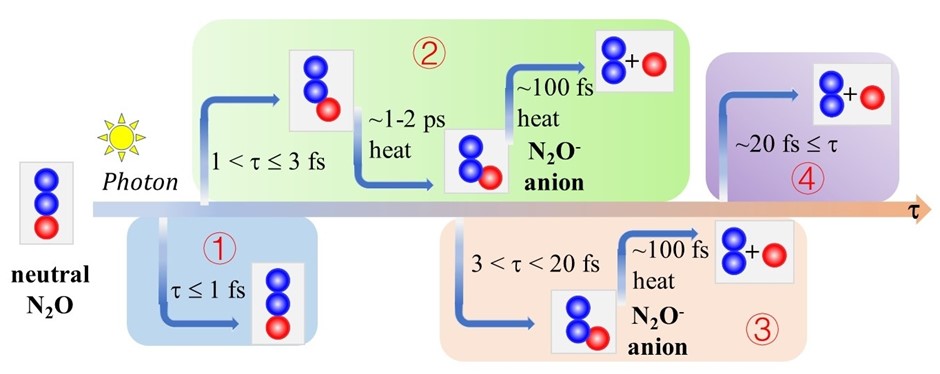
图5. 光催化及光热协同催化N2O解离示意图。
基于以上的计算结果,我们提出通过高价金属掺杂,增强p-d杂化,促使更多的电子转移至N2O,实现低温下N2O高效热解离(图5)所示。晶体轨道布局分析也表明,金属掺杂弱化N-O化学键,使得N-O的强度小于 = 2 fs时的强度。为了证实金属掺杂的结果,以W掺杂作为特例进行了分子动力学模拟,结果表明经过~2.2 ps的热活化,N2O中N-O键自发断裂,分子解离。以上的探究表明即使在没有光照的协助下,高价过渡金属也可以促进N2O解离,为实验提供了调控电子转移和化学反应的新策略。
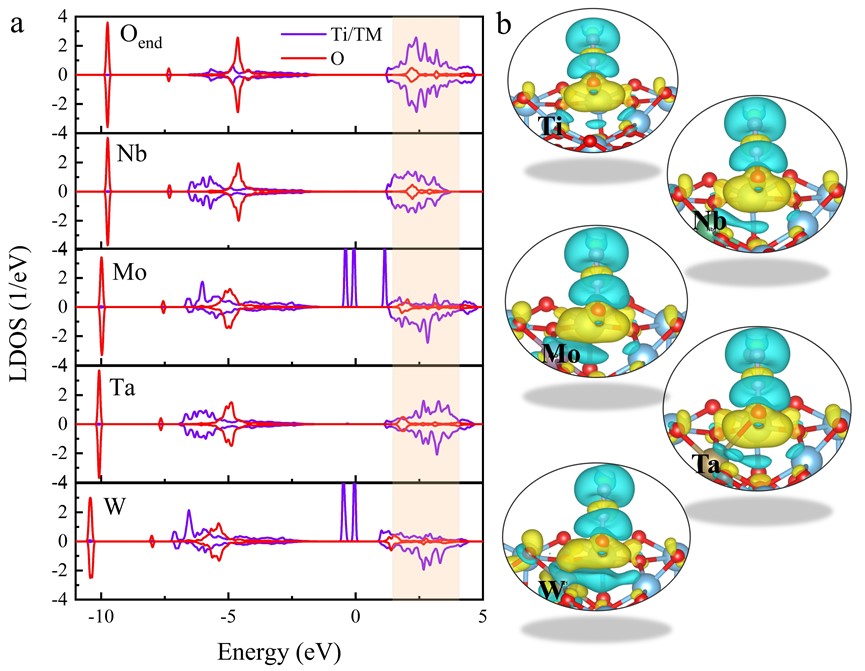
图6. 金属掺杂增强p-d杂化,实现N2O分子有效热解离。
上述研究成果“Photolysis versus Photothermolysis of N2O on a Semiconductor Surface Revealed by Nonadiabatic Molecular Dynamics”近期发表在The Journal of American Chemical Society(https://doi.org/10.1021/jacs.2c10643)。