吲哚大环阻转异构体结构单元是多个复杂天然产物的核心骨架,如cihunamide B和dynobactin A等。该类分子具有独特的3D结构及构象张力,是研究蛋白-蛋白相互作用中常用的结构。然而,该类分子在商业化小分子化合物库中鲜有存在,其可能原因为缺乏高效的合成方法。目前,底物诱导的非对映选择性大环化反应是构建吲哚大环阻转异构体的常用方法。仅有一例催化对映选择性构建该类结构的报道,即清华大学汪舰教授发展的氮杂环卡宾(NHC)催化的大环内酯化反应(Angew. Chem. Int. Ed. 2024, 63, e202316739)。(图一)
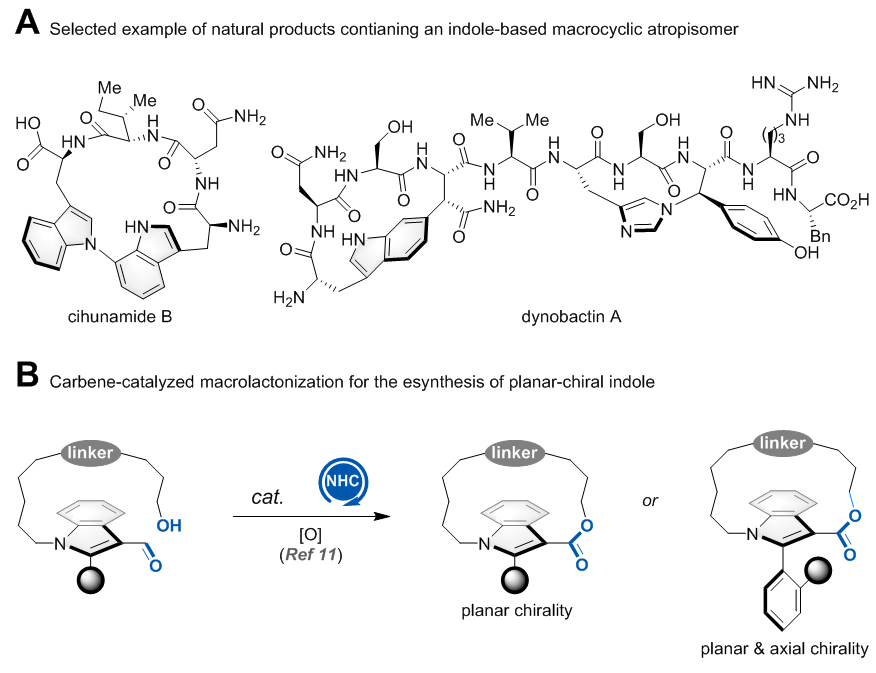
图一. 含吲哚大环阻转异构体的天然产物举例及NHC催化的大环内酯化反应
过渡金属催化的不对称吲哚合成法(如Cacchi反应、烯胺氮芳基化反应、Larock吲哚合成、Rh催化的C–H活化/环合反应等)受到了广泛关注。然而,不对称吲哚合成法大都集中于构建轴手性吲哚,尚未有利用这类方法构建大环阻转异构体的报道。不对称大环化反应在构建平面手性大环分子方面受到了关注,该类反应同时构建大环并控制平面手性,大大提高了合成效率。近年来,合成化学家先后发展了手性磷酸催化的羟基对联烯加成、酶催化的大环内酯化、氮杂环卡宾催化的大环内酯化、相转移催化的芳香亲核取代和钯催化的碳氧键偶联等不对称大环反应,这些开创性的方法为催化不对称构建平面手性环蕃烷提供了新思路。目前,这类策略以形成C–O键和C–N键为主,导致无法合成全碳链的平面手性大环结构。
近日,北京师范大学化学学院赵常贵课题组和济宁学院刘涛教授、吕康副教授团队合作发展了Rh催化的C–H活化/分子内氧化环化反应,高立体选择性地构建了吲哚大环阻转异构体。该反应利用手性螺环铑催化剂(J. Am. Chem. Soc. 2016, 138, 5242–5245),一步构建了大环和吲哚,并控制了平面手性,彰显了手性螺环铑催化剂的合成魅力。特别是,该反应可构建极具合成挑战性的全碳链平面手性大环。此外,作者系统研究了吲哚取代基、ansa链长度和取代基等因素对平面手性大环构象稳定性的影响。(图二)

图二. 过渡金属催化的不对称吲哚合成法构建大环阻转异构体
济宁学院刘涛教授和吕康副教授课题组对平面手性大环的构象稳定性及反应对映选择性产生的根源进行了DFT计算研究。结果表明,随着ansa链长度的增加,大环围绕平面翻转需要克服的能垒降低,翻转过程变得容易,这与实验测得的ee值一致,说明ansa链长度是影响构象稳定性的重要因素。此外,研究还发现烷基链中的取代基也可以影响构象稳定性。对反应机理的研究表明,反应经历了典型的Rh(III)催化C–H活化/氧化环化机理路径。其中,C–H活化及随后的分子内炔烃迁移插入共同决定了产物的对映选择性。
此工作得到了国家自然科学基金、山东省自然科学基金、济宁学院研究基金以及中央高校基本科研业务费专项资金的资助。相关成果以“Rhodium(III)-Catalyzed Atroposelective Indolization to Access Planar-Chiral Macrocycles”为题发表于《Journal of the American Chemical Society》杂志。北京师范大学化学学院为第一完成单位,北京师范大学硕士研究生翟泓轩、济宁学院吕康副教授和北京师范大学博士研究生李佳燕为论文的共同第一作者,济宁学院刘涛教授和北京师范大学赵常贵副教授为共同通讯作者。